 These pages are under construction, again.
These pages are under construction, again.
These pages are under construction, again.
Electronic structure studies
Electronic effects have fascinated physical organic chemists for many years, and for many problems empirical
rules or models have been developed. For example, in aromatic compounds, Hammett (and related) substituent
constants describe the effect of substituents on group properties of other, remote substituents
on the ring, such as acidity or hydrogen bond donating capacity,
or atomic properties, such as nucleophilicity, electrophilicity, chemical shift and more.
With the advent of highly sophisticated computational methods, empirical rules or models can finally be
confirmed and better understood, or, on occasion, be shown to be incomplete or even wrong. In fact, often
there is no longer any need to resort to the empirical rules or models of old.
Summary of selected studies
- Carbene character in nitrilimines:
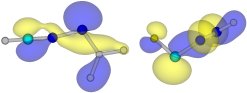
|
The HOMO of two nitrilimines, H-CNN-BH2 and F-CNN-H. N-BH2 substitution leads to predominantly 1,3-dipolar character with almost no carbenic contribution. C-F substitution allows stabilization of the C atom and brings forward the carbenic contribution. R. Mawhinney, 2004 |
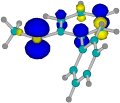 |
Spin density in the 2-methylthiobiphenyl radical cation, mostly on S. The twist in 2-methylthiobiphenyl is comparable and much smaller than for 2-t-butyl substitution. For t-butyl, the rings are oxidized instead, and the radical cation becomes more planar than for SMe. L. Zhang, 2010, PhD candidate |
| Return to |
H.M. Muchall HomePage Centre for Research in Molecular Modeling - CERMM |
Last update: august10; hmm